R/LinkedCharts Tutorial
Augmenting and checking a standard RNA-Seq analysis
Appendix
This document describes a standard analysis of the RNA-Seq dataset used in this tutorial.
We are working with data from this paper:
| C. Conway et al.: Elucidating drivers of oral epithelial dysplasia formation and malignant transformation to cancer using RNAseq. Oncotarget, 6:40186-40201 (2015), doi:10.18632/oncotarget.5529 |
Conweay et al. have collected tissue samples from 19 patients with oral squamous cell carcinoma. From each patient, they took 3 samples, one of normal oral mucosa (“N”), one of epithelial dysplasia (i.e., abnormal but not yet malignant tissue, “D”), and one sample of the tumour (“T”). They prepared RNA-Seq libraries from the samples and sequenced them. They deposited their raw data at the European Read Archive (ERA) under accession PRJEB7455 (secondary accession: ERP007185).
The Recount2 project (Collado Torres et al., Nature Biotechnology, 2017, doi:10.1038/nbt.3838) has downloaded this data set (and thousands other RNA-Seq data sets), ran it through a standardized preprocessing pipeline and provides a gene count table for download from the Recount2 web site.
Getting data and metadata
We download this count matrix from Recount2 (available via the secondary accession ERP007185) and read it into R
download.file("http://duffel.rail.bio/recount/v2/ERP007185/counts_gene.tsv.gz",
"ERP007185_counts_gene.tsv.gz" )
countMatrix <- as.matrix( read.table(
gzfile("ERP007185_counts_gene.tsv.gz"), header=TRUE, row.names="gene_id" ) )
The upper-left corner of the matrix looks like this
countMatrix[1:5,1:5]
## ERR649035 ERR649053 ERR649022 ERR649026 ERR649029
## ENSG00000000003.14 37354 136965 96040 144832 261208
## ENSG00000000005.5 371 119 1100 1139 1635
## ENSG00000000419.12 55566 53733 29553 111980 54818
## ENSG00000000457.13 77985 73875 44700 89683 83028
## ENSG00000000460.16 25159 31507 20118 37592 38827
The column names are ERA run accessions, whcih we first need to map to useful sample names. Unfortuantely, the sample table available from Recount2 does not contain a column that would actually describe the samples, the more complete sample table available from ERA, however, does (though it took us a bit to find this).
We went to the ERA page for the project. There, we clicked on “Select column” and selected the columns “Run accession” and “Submitter’s sample name”, then downloaded this table. A copy of this table is available from the present site: PRJEB7455.txt.
We will use tidyverse functionality to process the table
library( tidyverse )
We read in the table
sampleTable <- read_tsv( "PRJEB7455.txt" )
## Parsed with column specification:
## cols(
## run_accession = col_character(),
## sample_alias = col_character()
## )
sampleTable
## # A tibble: 57 x 2
## run_accession sample_alias
## <chr> <chr>
## 1 ERR649018 PG038-D
## 2 ERR649019 PG136-N
## 3 ERR649020 PG049-T
## 4 ERR649021 PG049-D
## 5 ERR649022 PG049-N
## 6 ERR649023 PG063-D
## 7 ERR649024 PG063-T
## 8 ERR649025 PG038-T
## 9 ERR649026 PG063-N
## 10 ERR649027 PG108-T
## # ... with 47 more rows
We split the sample name into its two parts, the patient ID (before the dash) and the tissue type (after the dash), then change the tissue type to more descriptive labels, impose an ordering of the levels by severity ( normal < dysplasia < tumour ), and finally sort the table.
sampleTable %>%
rename( sample_name = sample_alias ) %>%
separate( sample_name, c( "patient", "tissue" ), "-", remove=FALSE ) %>%
mutate( tissue = fct_recode( tissue, "normal"="N", "dysplasia"="D", "tumour"="T" ) ) %>%
mutate( tissue = fct_relevel( tissue, "normal", "dysplasia", "tumour" ) ) %>%
arrange( patient, tissue ) ->
sampleTable
This is now our final sample table
sampleTable
## # A tibble: 57 x 4
## run_accession sample_name patient tissue
## <chr> <chr> <chr> <fct>
## 1 ERR649059 PG004-N PG004 normal
## 2 ERR649060 PG004-D PG004 dysplasia
## 3 ERR649061 PG004-T PG004 tumour
## 4 ERR649035 PG038-N PG038 normal
## 5 ERR649018 PG038-D PG038 dysplasia
## 6 ERR649025 PG038-T PG038 tumour
## 7 ERR649022 PG049-N PG049 normal
## 8 ERR649021 PG049-D PG049 dysplasia
## 9 ERR649020 PG049-T PG049 tumour
## 10 ERR649026 PG063-N PG063 normal
## # ... with 47 more rows
Now, we bring the count matrix columns into the same order as in the sample table and replace the run accessions in the column names by the more readable sample names
countMatrix <- countMatrix[ , sampleTable$run_accession ]
colnames(countMatrix) <- sampleTable$sample_name
Next, we replace the rownames, currently Ensembl gene IDs, with gene names. To this, we first download from Ensembl Biomart a table mapping all Ensembl human gene IDs to gene symbols
bmtbl <- biomaRt::getBM(
c( "ensembl_gene_id", "external_gene_name" ),
mart = biomaRt::useEnsembl( "ensembl", "hsapiens_gene_ensembl" ) )
head(bmtbl)
## ensembl_gene_id external_gene_name
## 1 ENSG00000284532 MIR4723
## 2 ENSG00000238933 RF00019
## 3 ENSG00000275693 RF02116
## 4 ENSG00000275451 MIR6085
## 5 ENSG00000222870 RNU6-1328P
## 6 ENSG00000252023 RNU6-581P
We replace the rownames with the gene symbols, unless the gene symbol is missing or duplicated. As usual in R, such simple operations look most complicated:
tibble( rowname = rownames(countMatrix) ) %>%
mutate( ensembl_gene_id = str_extract( rowname, "ENSG\\d*" ) ) %>%
left_join( bmtbl, by = "ensembl_gene_id" ) %>%
mutate( new_rowname =
ifelse( ! is.na(external_gene_name) &
! external_gene_name %in% external_gene_name[duplicated(external_gene_name)],
external_gene_name,
rowname ) ) %>%
pull( new_rowname ) ->
rownames(countMatrix)
Now, we have this
countMatrix[ 1:5, 1:5 ]
## PG004-N PG004-D PG004-T PG038-N PG038-D
## TSPAN6 11642 25423 1526 37354 30699
## TNMD 405 0 1628 371 0
## DPM1 21828 32694 973 55566 33814
## SCYL3 31332 38436 11661 77985 63853
## C1orf112 14207 21808 8047 25159 25862
Running limma-voom
With everything in place, we now run a standard differential expression analysis using limma-voom (Law et al., Genome Biology, 2014, doi:10.1186/gb-2014-15-2-r29).
library( limma )
We aim to find any genes which differ significantly between any of the three tissue types, accounting for the patient as a blocking factor.
This is our model matrix
mm <- model.matrix( ~ patient + tissue, sampleTable )
colnames(mm)
## [1] "(Intercept)" "patientPG038" "patientPG049"
## [4] "patientPG063" "patientPG079" "patientPG086"
## [7] "patientPG105" "patientPG108" "patientPG114"
## [10] "patientPG122" "patientPG123" "patientPG129"
## [13] "patientPG136" "patientPG137" "patientPG144"
## [16] "patientPG146" "patientPG174" "patientPG187"
## [19] "patientPG192" "tissuedysplasia" "tissuetumour"
The model matrix columns concerning the tissue type are the last two
mm_tissue_columns <- which( attr( mm, "assign" ) == 2 )
mm_tissue_columns
## [1] 20 21
Now, we run the standard workflow of voom and limma
vm <- voom( countMatrix, mm )
fit <- lmFit( vm, mm )
fit <- eBayes( fit, robust=TRUE )
Last, we extract the results for an F test comparing the full model with the one missing the last two columns, to see which genes differ significantly between tissues.
voomResult <- topTable( fit, coef = mm_tissue_columns, number=Inf, sort.by="none" )
head(voomResult)
## tissuedysplasia tissuetumour AveExpr F P.Value
## TSPAN6 -0.6794364 -1.17596765 3.940362 17.369998 2.751020e-06
## TNMD -2.2216329 0.29408807 -4.690724 4.164227 2.206521e-02
## DPM1 0.3353803 0.14224571 3.551261 1.659938 2.018351e-01
## SCYL3 0.2533487 0.06609826 4.212959 2.152911 1.282229e-01
## C1orf112 0.3422943 0.32698821 3.350802 3.262281 4.774102e-02
## FGR 0.6687112 1.06189945 2.673165 7.042142 2.221402e-03
## adj.P.Val
## TSPAN6 8.515251e-05
## TNMD 7.345409e-02
## DPM1 3.391010e-01
## SCYL3 2.492604e-01
## C1orf112 1.264950e-01
## FGR 1.397246e-02
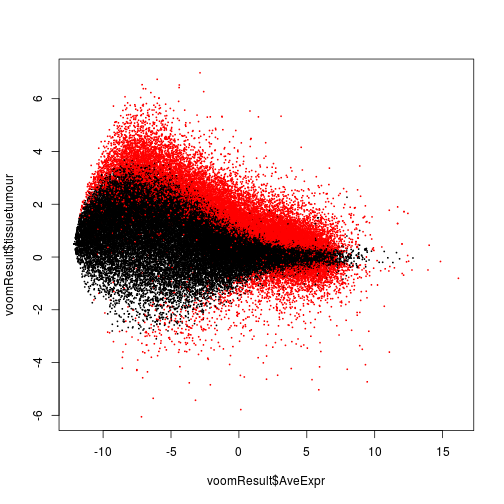
Let’s make an MA plot of these, showing the log fold change tumour/normal versus the average expression and highlighting genes significant at 10% FDR in the F test.
plot( voomResult$AveExpr, voomResult$tissuetumour,
col = ifelse( voomResult$adj.P.Val < .1, "red", "black" ), pch=20, cex=.3 )
Save the results
We save the result
save( countMatrix, sampleTable, voomResult, file="orcc.rda" )
This file is available here: orcc.rda
Session info
sessionInfo()
## R version 3.4.2 (2017-09-28)
## Platform: x86_64-pc-linux-gnu (64-bit)
## Running under: Ubuntu 16.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/openblas-base/libblas.so.3
## LAPACK: /usr/lib/libopenblasp-r0.2.18.so
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] limma_3.34.9 bindrcpp_0.2.2 forcats_0.3.0 stringr_1.3.1
## [5] dplyr_0.7.6 purrr_0.2.5 readr_1.1.1 tidyr_0.8.1
## [9] tibble_1.4.2 ggplot2_3.0.0 tidyverse_1.2.1
##
## loaded via a namespace (and not attached):
## [1] Rcpp_0.12.18 lubridate_1.7.4 lattice_0.20-35
## [4] prettyunits_1.0.2 assertthat_0.2.0 digest_0.6.17
## [7] utf8_1.1.4 R6_2.2.2 cellranger_1.1.0
## [10] plyr_1.8.4 backports_1.1.2 stats4_3.4.2
## [13] RSQLite_2.1.1 evaluate_0.11 highr_0.7
## [16] httr_1.3.1 pillar_1.3.0 rlang_0.2.2
## [19] progress_1.2.0 lazyeval_0.2.1 curl_3.2
## [22] readxl_1.1.0 rstudioapi_0.7 blob_1.1.1
## [25] S4Vectors_0.16.0 statmod_1.4.30 RCurl_1.95-4.11
## [28] bit_1.1-14 biomaRt_2.34.2 munsell_0.5.0
## [31] broom_0.5.0 compiler_3.4.2 modelr_0.1.2
## [34] pkgconfig_2.0.2 BiocGenerics_0.24.0 tidyselect_0.2.4
## [37] IRanges_2.12.0 XML_3.98-1.16 fansi_0.3.0
## [40] crayon_1.3.4 withr_2.1.2 bitops_1.0-6
## [43] grid_3.4.2 nlme_3.1-137 jsonlite_1.5
## [46] gtable_0.2.0 DBI_1.0.0 magrittr_1.5
## [49] scales_1.0.0 cli_1.0.0 stringi_1.2.4
## [52] xml2_1.2.0 org.Hs.eg.db_3.5.0 tools_3.4.2
## [55] bit64_0.9-7 Biobase_2.38.0 glue_1.3.0
## [58] hms_0.4.2 parallel_3.4.2 AnnotationDbi_1.40.0
## [61] colorspace_1.3-2 rvest_0.3.2 memoise_1.1.0
## [64] knitr_1.20 bindr_0.1.1 haven_1.1.2